Characterization of a New Member of Alphacoronavirus with Unique Genomic Features in Rhinolophus Bats


Abstract
:1. Introduction
2. Materials and Methods
2.1. Ethics Statement
2.2. Sampling
2.3. RNA Extraction, PCR Screening and Sequencing
2.4. Sequencing of Full-Length Genomes
2.5. Genome Analysis
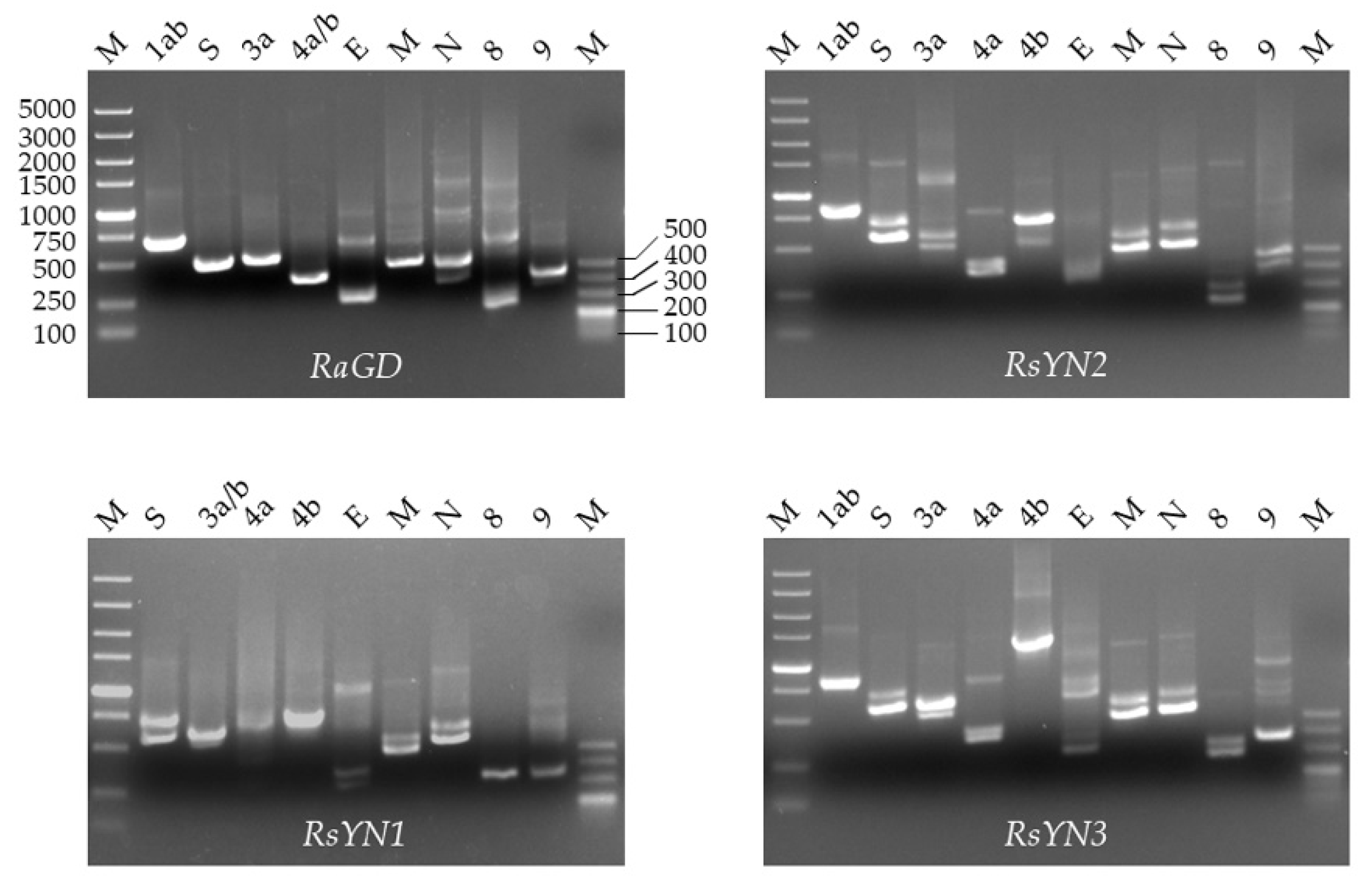
2.6. Transcriptional Analysis of Subgenomic mRNA
2.7. Cell Lines, Gene Cloning, and Expression
2.8. Virus Isolation
2.9. Apoptosis Analysis
2.10. Dual Luciferase Reporter Assays
2.11. BtCoV/Rh/YN2012 Spike-Mediated Pseudoviruses Cell Tropism Screening
2.12. Nucleotide Sequence Accession Numbers
3. Results
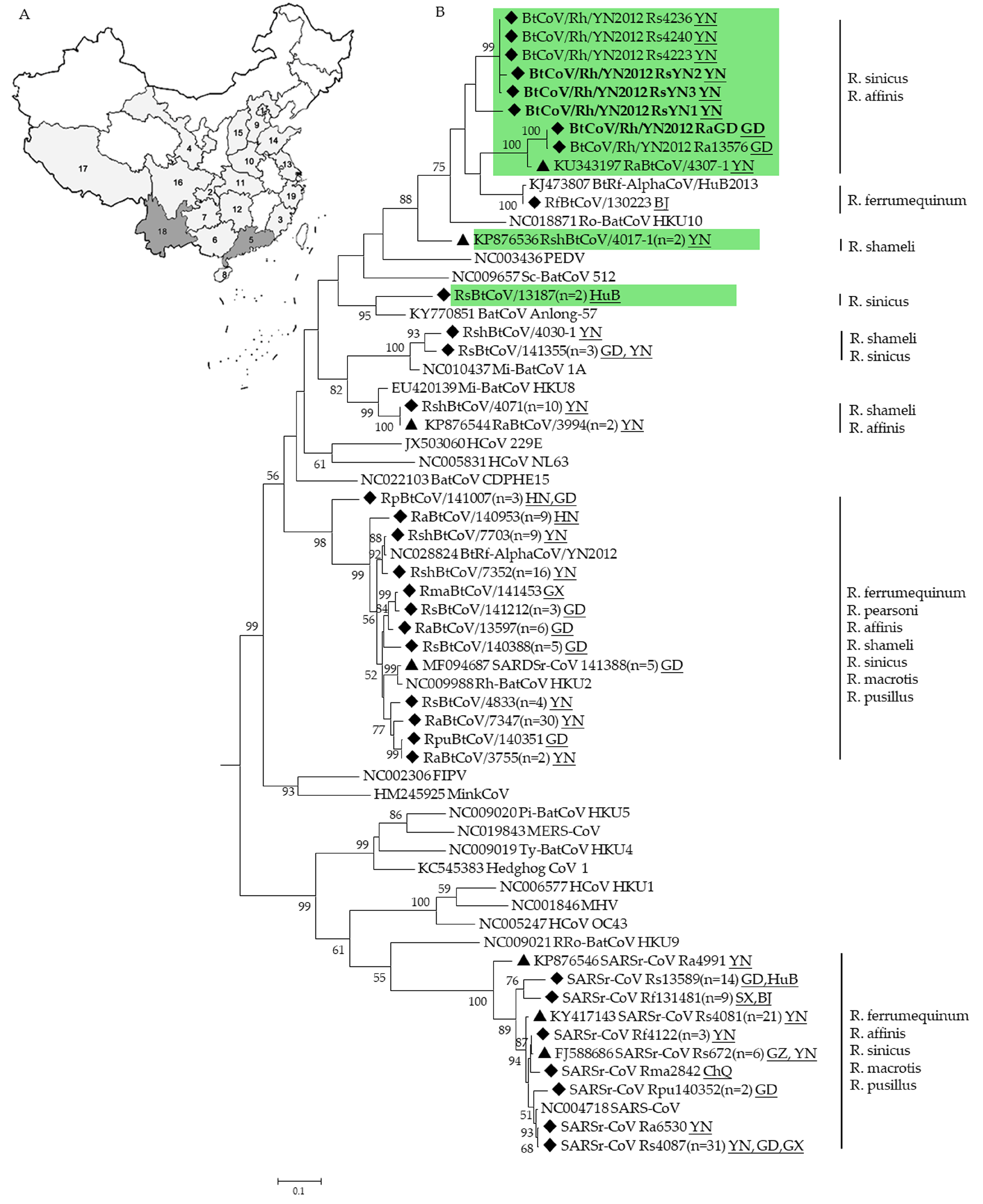
3.1. CoVs Detected in Rhinolophus Bats
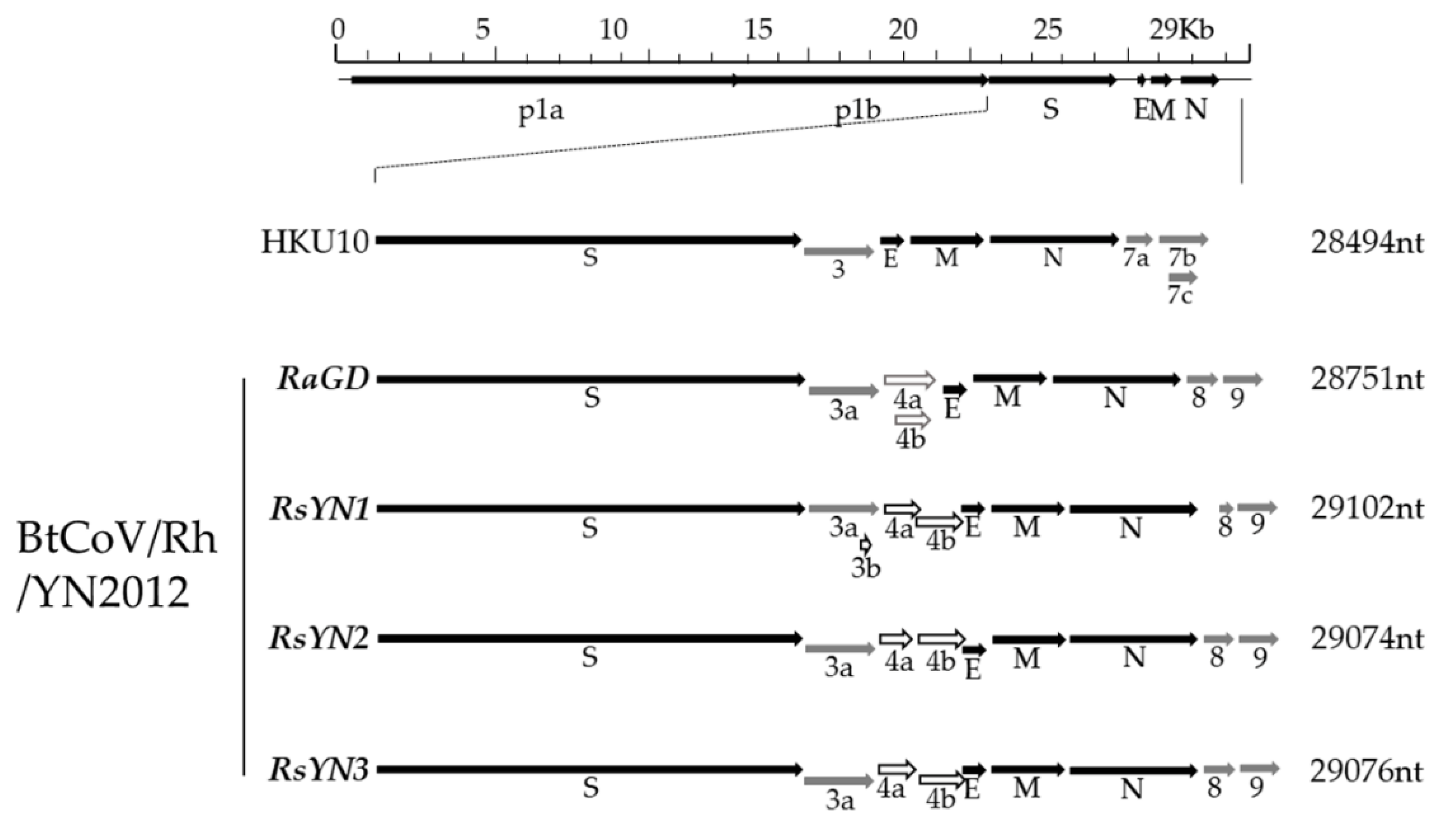
3.2. Genomic Characterization of a Novel Alpha-CoV (BtCoV/Rh/YN2012)
3.3. Subgenomic Structures and Accessory Genes of BtCoV/Rh/YN2012
3.4. Phylogenetic Analysis
3.5. Estimation of Synonymous and Nonsynonymous Substitution Rates
3.6. Apoptosis Analysis of ORF9
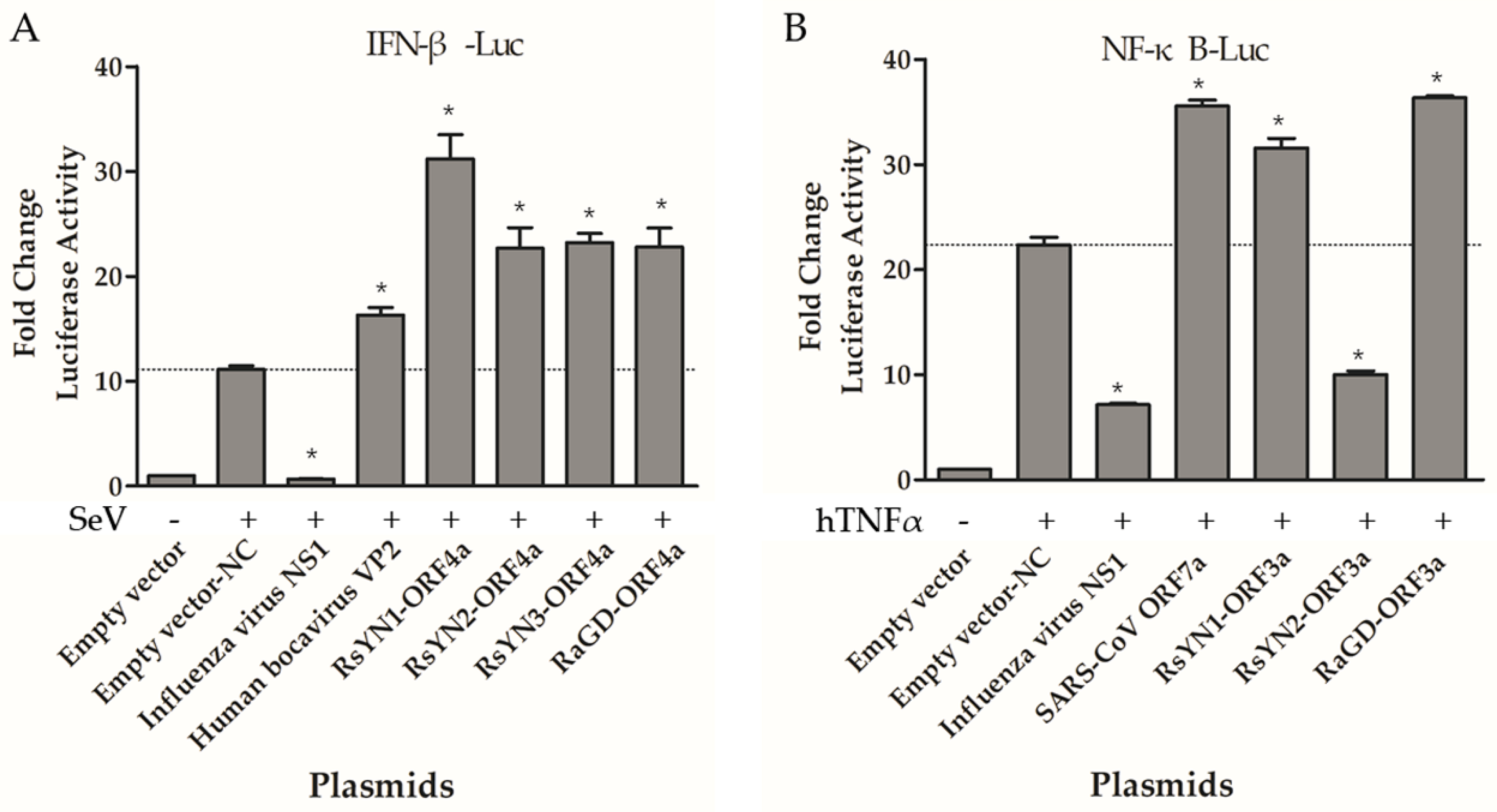
3.7. ORF4a Proteins Induce Production of IFN-β
3.8. ORF3a Proteins Modulate NF-κB
3.9. BtCoV/Rh/YN2012 Spike Mediated Pseudovirus Entry
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- David, M.; Knipe, P.M.H. Fields Virology; Lippincott Williams and Wilkins: Philadelphia, PA, USA, 2013. [Google Scholar]
- King, A.M.Q.; Lefkowitz, E.J.; Mushegian, A.R.; Adams, M.J.; Dutilh, B.E.; Gorbalenya, A.E.; Harrach, B.; Harrison, R.L.; Junglen, S.; Knowles, N.J.; et al. Changes to taxonomy and the international code of virus classification and nomenclature ratified by the international committee on taxonomy of viruses (2018). Arch. Virol. 2018, 163, 2601–2631. [Google Scholar] [CrossRef]
- Virus Taxonomy: 2018b Release. Available online: Https://viralzone.Expasy.Org/785 (accessed on 7 April 2019).
- Woo, P.C.Y.; Lau, S.K.P.; Lam, C.S.F.; Lau, C.C.Y.; Tsang, A.K.L.; Lau, J.H.N.; Bai, R.; Teng, J.L.L.; Tsang, C.C.C.; Wang, M.; et al. Discovery of seven novel mammalian and avian coronaviruses in the genus deltacoronavirus supports bat coronaviruses as the gene source of alphacoronavirus and betacoronavirus and avian coronaviruses as the gene source of gammacoronavirus and deltacoronavirus. J. Virol. 2012, 86, 3995–4008. [Google Scholar]
- Woo, P.C.Y.; Lau, S.K.P.; Lam, C.S.F.; Tsang, A.K.L.; Hui, S.W.; Fan, R.Y.Y.; Martelli, P.; Yuen, K.Y. Discovery of a novel bottlenose dolphin coronavirus reveals a distinct species of marine mammal coronavirus in gammacoronavirus. J. Virol. 2014, 88, 1318–1331. [Google Scholar] [CrossRef] [PubMed]
- Hu, B.; Ge, X.Y.; Wang, L.F.; Shi, Z.L. Bat origin of human coronaviruses. Virol. J. 2015, 12. [Google Scholar] [CrossRef] [PubMed]
- Jung, K.; Saif, L.J. Porcine epidemic diarrhea virus infection: Etiology, epidemiology, pathogenesis and immunoprophylaxis. Vet. J. 2015, 204, 134–143. [Google Scholar] [CrossRef]
- Gong, L.; Li, J.; Zhou, Q.F.; Xu, Z.C.; Chen, L.; Zhang, Y.; Xue, C.Y.; Wen, Z.F.; Cao, Y.C. A new bat-hku2-like coronavirus in swine, China, 2017. Emerg. Infect. Dis 2017, 23, 1607–1609. [Google Scholar] [CrossRef]
- Pan, Y.F.; Tian, X.Y.; Qin, P.; Wang, B.; Zhao, P.W.; Yang, Y.L.; Wang, L.X.; Wang, D.D.; Song, Y.H.; Zhang, X.B.; et al. Discovery of a novel swine enteric alphacoronavirus (SeACoV) in southern China. Vet. Microbiol. 2017, 211, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Chae, C.; Kim, O.; Choi, C.; Min, K.; Cho, W.S.; Kim, J.; Tai, J.H. Prevalence of porcine epidemic diarrhoea virus and transmissible gastroenteritis virus infection in korean pigs. Vet. Record 2000, 147, 606–608. [Google Scholar] [CrossRef]
- Wu, Z.Q.; Yang, L.; Ren, X.W.; He, G.M.; Zhang, J.P.; Yang, J.; Qian, Z.H.; Dong, J.; Sun, L.L.; Zhu, Y.F.; et al. Deciphering the bat virome catalog to better understand the ecological diversity of bat viruses and the bat origin of emerging infectious diseases. Isme J. 2016, 10, 609–620. [Google Scholar] [CrossRef] [PubMed]
- Ge, X.Y.; Li, J.L.; Yang, X.L.; Chmura, A.A.; Zhu, G.J.; Epstein, J.H.; Mazet, J.K.; Hu, B.; Zhang, W.; Peng, C.; et al. Isolation and characterization of a bat SARS-like coronavirus that uses the ACE2 receptor. Nature 2013, 503, 535–538. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, X.L.; Hu, B.; Wang, B.; Wang, M.N.; Zhang, Q.; Zhang, W.; Wu, L.J.; Ge, X.Y.; Zhang, Y.Z.; Daszak, P.; et al. Isolation and characterization of a novel bat coronavirus closely related to the direct progenitor of severe acute respiratory syndrome coronavirus. J. Virol. 2016, 90, 3253–3256. [Google Scholar] [CrossRef]
- Menachery, V.D.; Yount, B.L.; Sims, A.C.; Debbink, K.; Agnihothram, S.S.; Gralinski, L.E.; Graham, R.L.; Scobey, T.; Plante, J.A.; Royal, S.R.; et al. Sars-like wiv1-cov poised for human emergence. Proc. Natl. Acad. Sci. USA. 2016, 113, 3048–3053. [Google Scholar] [CrossRef]
- Zhou, P.; Fan, H.; Lan, T.; Yang, X.-L.; Shi, W.-F.; Zhang, W.; Zhu, Y.; Zhang, Y.-W.; Xie, Q.-M.; Mani, S.; et al. Fatal swine acute diarrhea syndrome caused by an HKU2-related coronavirus of bat origin. Nature 2018, 556, 255–258. [Google Scholar] [CrossRef]
- Li, W.D.; Shi, Z.L.; Yu, M.; Ren, W.Z.; Smith, C.; Epstein, J.H.; Wang, H.Z.; Crameri, G.; Hu, Z.H.; Zhang, H.J.; et al. Bats are natural reservoirs of sars-like coronaviruses. Science 2005, 310, 676–679. [Google Scholar] [CrossRef]
- Luna, L.K.D.; Heiser, V.; Regamey, N.; Panning, M.; Drexler, J.F.; Mulangu, S.; Poon, L.; Baumgarte, S.; Haijema, B.J.; Kaiser, L.; et al. Generic detection of coronaviruses and differentiation at the prototype strain level by reverse transcription-PCR and nonfluorescent low-density microarray. J. Clin. Microbiol. 2007, 45, 1049–1052. [Google Scholar] [CrossRef]
- Hu, B.; Zeng, L.P.; Yang, X.L.; Ge, X.Y.; Zhang, W.; Li, B.; Xie, J.Z.; Shen, X.R.; Zhang, Y.Z.; Wang, N.; et al. Discovery of a rich gene pool of bat sars-related coronaviruses provides new insights into the origin of SARS coronavirus. PLOS Pathog. 2017, 13, e1006698. [Google Scholar] [CrossRef] [PubMed]
- Irwin, D.M.; Kocher, T.D.; Wilson, A.C. Evolution of the cytochrome-b gene of mammals. J. Mol. Evol. 1991, 32, 128–144. [Google Scholar] [CrossRef]
- Mayer, F.; von Helversen, O. Cryptic diversity in european bats. Proc. Biol. Sci. 2001, 268, 1825–1832. [Google Scholar] [CrossRef] [PubMed]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C.; et al. Geneious basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef]
- Yang, D.; Leibowitz, J.L. The structure and functions of coronavirus genomic 3' and 5' ends. Virus Res. 2015, 206, 120–133. [Google Scholar] [CrossRef] [PubMed]
- Tamura, K.; Stecher, G.; Peterson, D.; Filipski, A.; Kumar, S. Mega6: Molecular evolutionary genetics analysis version 6.0. Mol. Biol. Evol. 2013, 30, 2725–2729. [Google Scholar] [CrossRef] [PubMed]
- Hall, B.G. Building phylogenetic trees from molecular data with MEGA. Mol. Biol. Evol. 2013, 30, 1229–1235. [Google Scholar] [CrossRef]
- Martin, D.P.; Lemey, P.; Lott, M.; Moulton, V.; Posada, D.; Lefeuvre, P. Rdp3: A flexible and fast computer program for analyzing recombination. Bioinformatics 2010, 26, 2462–2463. [Google Scholar] [CrossRef] [PubMed]
- Lole, K.S.; Bollinger, R.C.; Paranjape, R.S.; Gadkari, D.; Kulkarni, S.S.; Novak, N.G.; Ingersoll, R.; Sheppard, H.W.; Ray, S.C. Full-length human immunodeficiency virus type 1 genomes from subtype C-infected seroconverters in India, with evidence of intersubtype recombination. J. Virol. 1999, 73, 152–160. [Google Scholar]
- Siltberg, J.; Liberles, D.A. A simple covarion-based approach to analyse nucleotide substitution rates. J. Evol. Biol. 2002, 15, 588–594. [Google Scholar] [CrossRef]
- Zimmermann, L.; Stephens, A.; Nam, S.Z.; Rau, D.; Kubler, J.; Lozajic, M.; Gabler, F.; Soding, J.; Lupas, A.N.; Alva, V. A completely reimplemented mpi bioinformatics toolkit with a new hhpred server at its core. J. Mol. Biol. 2018, 430, 2237–2243. [Google Scholar] [CrossRef]
- Huang, C.; Liu, W.J.; Xu, W.; Jin, T.; Zhao, Y.; Song, J.; Shi, Y.; Ji, W.; Jia, H.; Zhou, Y.; et al. A bat-derived putative cross-family recombinant coronavirus with a reovirus gene. PLOS Pathog. 2016, 12, e1005883. [Google Scholar] [CrossRef] [PubMed]
- Zeng, L.P.; Gao, Y.T.; Ge, X.Y.; Zhang, Q.; Peng, C.; Yang, X.L.; Tan, B.; Chen, J.; Chmura, A.A.; Daszak, P.; et al. Bat severe acute respiratory syndrome-like coronavirus wiv1 encodes an extra accessory protein, ORFX, involved in modulation of the host immune response. J. Virol. 2016, 90, 6573–6582. [Google Scholar] [CrossRef]
- Zhou, P.; Li, H.X.; Wang, H.Z.; Wang, L.F.; Shi, Z.L. Bat severe acute respiratory syndrome-like coronavirus ORF3B homologues display different interferon antagonist activities. J. Gen. Virol. 2012, 93, 275–281. [Google Scholar] [CrossRef]
- Wang, X.Y.; Li, M.; Zheng, H.Y.; Muster, T.; Palese, P.; Beg, A.A.; Garcia-Sastre, A. Influenza a virus NS1 protein prevents activation of NF-kappa B and induction of alpha/beta interferon. J. Virol. 2000, 74, 11566–11573. [Google Scholar] [CrossRef]
- Kanzawa, N.; Nishigaki, K.; Hayashi, T.; Ishii, Y.; Furukawa, S.; Niiro, A.; Yasui, F.; Kohara, M.; Morita, K.; Matsushima, K.; et al. Augmentation of chemokine production by severe acute respiratory syndrome coronavirus 3a/X1 and 7a/X4 proteins through NF-kappa B activation. FEBS Lett. 2006, 580, 6807–6812. [Google Scholar] [CrossRef]
- Luo, H.L.; Zhang, Z.F.; Zheng, Z.H.; Ke, X.L.; Zhang, X.W.; Li, Q.; Liu, Y.; Bai, B.K.; Mao, P.Y.; Hu, Q.X.; et al. Human bocavirus VP2 upregulates IFN-beta pathway by inhibiting ring finger protein 125-mediated ubiquitination of retinoic acid-inducible gene-I. J. Immunol. 2013, 191, 660–669. [Google Scholar] [CrossRef] [PubMed]
- Luo, C.-M.; Wang, N.; Yang, X.-L.; Liu, H.-Z.; Zhang, W.; Li, B.; Hu, B.; Peng, C.; Geng, Q.-B.; Zhu, G.-J.; et al. Discovery of novel bat coronaviruses in south china that use the same receptor as MERS coronavirus. J. Virol. 2018, 92. [Google Scholar] [CrossRef]
- Yang, Y.; Du, L.Y.; Liu, C.; Wang, L.L.; Ma, C.Q.; Tang, J.; Baric, R.S.; Jiang, S.; Li, F. Receptor usage and cell entry of bat coronavirus hku4 provide insight into bat-to-human transmission of mers coronavirus. Proc. Natl. Acad. Sci. USA 2014, 111, 12516–12521. [Google Scholar] [CrossRef] [PubMed]
- Chu, D.K.W.; Peiris, J.S.M.; Chen, H.; Guan, Y.; Poon, L.L.M. Genomic characterizations of bat coronaviruses (1A, 1B and HKU8) and evidence for co-infections in miniopterus bats. J. Gen. Virol. 2008, 89, 1282–1287. [Google Scholar] [CrossRef]
- Marra, M.A.; Jones, S.J.M.; Astell, C.R.; Holt, R.A.; Brooks-Wilson, A.; Butterfield, Y.S.N.; Khattra, J.; Asano, J.K.; Barber, S.A.; Chan, S.Y.; et al. The genome sequence of the SARS-associated coronavirus. Science 2003, 300, 1399–1404. [Google Scholar] [CrossRef]
- Lau, S.K.P.; Woo, P.C.Y.; Li, K.S.M.; Huang, Y.; Wang, M.; Lam, C.S.F.; Xu, H.F.; Guo, R.T.; Chan, K.H.; Zheng, B.J.; et al. Complete genome sequence of bat coronavirus HKU2 from chinese horseshoe bats revealed a much smaller spike gene with a different evolutionary lineage from the rest of the genome. Virology 2007, 367, 428–439. [Google Scholar] [CrossRef] [PubMed]
- Ge, X.Y.; Wang, N.; Zhang, W.; Hu, B.; Li, B.; Zhang, Y.Z.; Zhou, J.H.; Luo, C.M.; Yang, X.L.; Wu, L.J.; et al. Coexistence of multiple coronaviruses in several bat colonies in an abandoned mineshaft. Virol. Sin. 2016, 31, 31–40. [Google Scholar] [CrossRef] [PubMed]
- Yuan, J.F.; Hon, C.C.; Li, Y.; Wang, D.M.; Xu, G.L.; Zhang, H.J.; Zhou, P.; Poon, L.L.M.; Lam, T.T.Y.; Leung, F.C.C.; et al. Intraspecies diversity of sars-like coronaviruses in Rhinolophus Sinicus and its implications for the origin of SARS coronaviruses in humans. J. Gen. Virol. 2010, 91, 1058–1062. [Google Scholar] [CrossRef]
- De Groot, R.J.; Baker, S.C.; Baric, R.; Enjuanes, L.; Gorbalenya, A.E.; Holmes, K.V.; Perlman, S.; Poon, L.; Rottier, P.J.M.; Talbot, P.J.; et al. Family Coronaviridae. In Virus Taxonomy, Classification and Nomenclature of Viruses. Ninth Report of the International Committee on Taxonomy of Viruses, 1st ed.; King, A.M.Q., Adams, M.J., Carstens, E.B., Lefkowitz, E.J., Eds.; Elsevier Academic Press: San Diego, CA, USA, 2011; pp. 806–828. [Google Scholar]
- Liu, D.X.; Fung, T.S.; Chong, K.K.L.; Shukla, A.; Hilgenfeld, R. Accessory proteins of SARS-CoV and other coronaviruses. Antiviral Res. 2014, 109, 97–109. [Google Scholar] [CrossRef]
- Chang, H.W.; de Groot, R.J.; Egberink, H.F.; Rottier, P.J.M. Feline infectious peritonitis: Insights into feline coronavirus pathobiogenesis and epidemiology based on genetic analysis of the viral 3C gene. J. Gen. Virol. 2010, 91, 415–420. [Google Scholar] [CrossRef]
- Chang, H.W.; Egberink, H.F.; Rottier, P.J.M. Sequence analysis of feline coronaviruses and the circulating virulent/avirulent theory. Emerg. Infect. Dis. 2011, 17, 744–746. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, N.C.; Liu, H.W.; Scarlett, J.; Leutenegger, C.M.; Golovko, L.; Kennedy, H.; Kamal, F.M. Feline infectious peritonitis: Role of the feline coronavirus 3c gene in intestinal tropism and pathogenicity based upon isolates from resident and adopted shelter cats. Virus Res. 2012, 165, 17–28. [Google Scholar] [CrossRef] [PubMed]
- De Haan, C.A.M.; Masters, P.S.; Shen, X.L.; Weiss, S.; Rottier, P.J.M. The group-specific murine coronavirus genes are not essential, but their deletion, by reverse genetics, is attenuating in the natural host. Virology 2002, 296, 177–189. [Google Scholar] [CrossRef] [PubMed]
- Karjee, S.; Minhas, A.; Sood, V.; Ponia, S.S.; Banerjea, A.C.; Chow, V.T.K.; Mukherjee, S.K.; Lal, S.K. The 7a accessory protein of severe acute respiratory syndrome coronavirus acts as an RNA silencing suppressor. J. Virol. 2010, 84, 10395–10401. [Google Scholar] [CrossRef] [PubMed]
- Kopecky-Bromberg, S.A.; Martinez-Sobrido, L.; Palese, P. 7a protein of severe acute respiratory syndrome coronavirus inhibits cellular protein synthesis and activates p38 mitogen-activated protein kinase. J. Virol. 2006, 80, 785–793. [Google Scholar] [CrossRef]
- Yuan, X.L.; Wu, J.; Shan, Y.J.; Yao, Z.Y.; Dong, B.; Chen, B.; Zhao, Z.H.; Wang, S.Q.; Chen, J.P.; Cong, Y.W. SARS coronavirus 7a protein blocks cell cycle progression at G0/G1 phase via the cyclin D3/pRb pathway. Virology 2006, 346, 74–85. [Google Scholar] [CrossRef] [Green Version]
- Tan, Y.J.; Fielding, B.C.; Goh, P.Y.; Shen, S.; Tan, T.H.P.; Lim, S.G.; Hong, W.J. Overexpression of 7a, a protein specifically encoded by the severe acute respiratory syndrome coronavirus, induces apoptosis via a caspase-dependent pathway. J. Virol. 2004, 78, 14043–14047. [Google Scholar] [CrossRef]
- Tan, Y.X.; Tan, T.H.P.; Lee, M.J.R.; Tham, P.Y.; Gunalan, V.; Druce, J.; Birch, C.; Catton, M.; Fu, N.Y.; Yu, V.C.; et al. Induction of apoptosis by the severe acute respiratory syndrome coronavirus 7a protein is dependent on its interaction with the Bcl-XL protein. J. Virol. 2007, 81, 6346–6355. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Bat Species | Samples | CoV Species Positive (Positive Rate %) | Province1 | ||||||
|---|---|---|---|---|---|---|---|---|---|
| HKU2 | HKU8 | HuB2013 | Mi-BtCoV 1 | SARSr-CoV | BtCoV/Rh/YN2012 | Other AlphaCoVs | |||
| R.affinis | 499 | 52(10.4) | 2(0.4) | 2(0.4) | 3(0.6) | 5, 7, 8, 10, 13, 18, 19 | |||
| R.blythi | 17 | 5,11,18 | |||||||
| R.ferrumequinum | 238 | 1(0.4) | 12(5.0) | 1,3,4,7,9,10,14,15,18 | |||||
| R.lepidus | 21 | 3 | |||||||
| R.luctus | 8 | 5,8,19 | |||||||
| R.macrotis | 31 | 1(3.2) | 1(3.2) | 2,5,6,9,11,13,1,18 | |||||
| R.monoceros | 5 | 18 | |||||||
| R.pearsoni | 106 | 3,5,6,7,10,17,18 | |||||||
| R.pusillus | 283 | 4(1.4) | 2(0.7) | 3,5,6,9,10,11,13,14 | |||||
| R.shamelli | 55 | 25(45.5) | 10(18.2) | 1(0.4) | 2(0.7)2 | 18 | |||
| R.sinicus | 740 | 12(1.6) | 3(0.4) | 72(9.7) | 6(0.8) | 2(0.3)3 | 3,5,6,7,8,11,12,13,16,18,19 | ||
| R.spp. | 58 | 2,6,7,18 | |||||||
| Total | 2061 | 94(4.6) | 12(0.6) | 1(0.05) | 4(0.2) | 89(4.3) | 9(0.4) | 4(0.2) | |
| CoV | ORFs | Nucleotide Positions | Predicted Size (AA) of Protein | Pairwise AA Identity (%) | TRS Sequence | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| RsYN1 | RsYN2 | RsYN3 | RaGD | HuB2013 | HKU10 | |||||
| RsYN1 | 1ab | 298–20465 | 6722 | 93.0 | 93.0 | 83.7 | 81.8 | 76.4 | CTAAAC(216)ATG | |
| S | 20472–24533 | 1353 | 68.4 | 67.9 | 64.9 | 62.3 | 61.7 | CTAAAT(8)ATG | ||
| S1 | 20541–22634 | 698 | 60.2 | 59.4 | 56.7 | 53.7 | 53.2 | |||
| S2 | 22659–24527 | 623 | 76.9 | 77.0 | 74.0 | 72.1 | 71.4 | |||
| NS3a | 24533–25201 | 222 | 65.0 | 65.0 | 54.3 | 52.4 | 53.5 | CAATAC(26)ATG | ||
| NS3b | 24978–25070 | 30 | NA | NA | NA | NA | NA | CCTTAC(35)ATG | ||
| NS4a | 25222–25581 | 119 | 47.1 | 62.2 | 18.6 | NA | NA | CTTTAC(50)ATG | ||
| NS4b | 25563–26009 | 148 | 57.8 | 52.9 | 11.4 | NA | NA | |||
| E | 25993–26217 | 74 | 89.2 | 89.2 | 77.1 | 68.7 | 75.9 | CTAAAC(66)ATG | ||
| M | 26229–26912 | 227 | 88.8 | 89.2 | 78.1 | 86.6 | 85.5 | CTAAAC(1)ATG | ||
| N | 26923–28134 | 403 | 84.3 | 84.3 | 68.1 | 70.3 | 61.8 | CTAAAC(3)ATG | ||
| NS8 | 28290–28409 | 39 | 26.7 | 33.3 | 28.6 | NA | NA | CTAAAC(0)ATG | ||
| NS9 | 28454–28819 | 121 | 78.1 | 50.6 | 55.6 | NA | NA | TTTCAC(3)ATG | ||
| Concatenated domains1 | 13,650 | 4550 | 97.3 | 97.4 | 91.9 | 84.2 | 86.9 | |||
| RsYN2 | 1ab | 298–20435 | 6712 | 93.0 | 99.4 | 84.4 | 82.5 | 76.9 | CTAAAC(216)ATG | |
| S | 20437–24495 | 1352 | 68.4 | 93.6 | 66.9 | 65.7 | 62.4 | CTAAAT(3)ATG | ||
| S1 | 20509–22599 | 697 | 60.2 | 88.2 | 58.7 | 58.8 | 55.6 | |||
| S2 | 22624–24489 | 623 | 76.9 | 99.9 | 76.3 | 72.8 | 70.0 | |||
| NS3a | 24495–25154 | 219 | 65.0 | 100.0 | 55.9 | 52.8 | 55.5 | CGTTAC(26)ATG | ||
| NS4a | 25164–25488 | 104 | 47.1 | 64.4 | 17.5 | NA | NA | CTTCAC(24)ATG | ||
| NS4b | 25507–25953 | 148 | 57.8 | 94.2 | 12.5 | NA | NA | |||
| E | 25934–26158 | 74 | 89.2 | 100.0 | 81.9 | 66.3 | 77.1 | CTAAAC(66)ATG | ||
| M | 26170–26859 | 229 | 84.3 | 99.8 | 79.9 | 90.0 | 88.8 | CTAAAC(1)ATG | ||
| N | 26870–28096 | 408 | 84.3 | 99.8 | 68.3 | 69.9 | 63.0 | CTAAAC(3)ATG | ||
| NS8 | 28107–28385 | 92 | 26.7 | 66.7 | 64.8 | NA | NA | CTAAAC(0)ATG | ||
| NS9 | 28425–28790 | 121 | 78.1 | 77.5 | 58.1 | NA | NA | TTTCAC(3)ATG | ||
| Concatenated domains | 13641 | 4547 | 97.3 | 99.6 | 91.8 | 84.3 | 87.1 | |||
| RsYN3 | 1ab | 298–20435 | 6712 | 93.0 | 99.4 | 84.4 | 82.5 | 76.9 | CTAAAC(216)ATG | |
| S | 20437–24495 | 1352 | 67.9 | 93.6 | 66.6 | 65.8 | 62.4 | CTAAAT(3)ATG | ||
| S1 | 20506–22599 | 698 | 59.4 | 88.2 | 57.9 | 59.2 | 54.9 | |||
| S2 | 22621–24489 | 623 | 77.0 | 99.9 | 76.4 | 72.9 | 70.1 | |||
| NS3a | 24495–25154 | 219 | 65.0 | 100.0 | 55.9 | 52.8 | 55.5 | CGTTAC(26)ATG | ||
| NS4a | 25164–25526 | 120 | 62.2 | 64.4 | 17.6 | NA | NA | CTTCAC(24)ATG | ||
| NS4b | 25508–25954 | 148 | 52.9 | 94.2 | 12.5 | NA | NA | |||
| E | 25935–26159 | 74 | 89.2 | 100.0 | 81.9 | 66.3 | 77.1 | CTAAAC(66)ATG | ||
| M | 26171–26860 | 229 | 89.2 | 99.6 | 80.3 | 90.3 | 89.2 | CTAAAC(1)ATG | ||
| N | 26871–28097 | 408 | 84.3 | 99.8 | 68.3 | 69.9 | 62.8 | CTAAAC(3)ATG | ||
| NS8 | 28108–28386 | 92 | 33.3 | 66.7 | 63.8 | NA | NA | CTAAAC(0)ATG | ||
| NS9 | 28434–28793 | 119 | 71.2 | 77.5 | 50.6 | NA | NA | TTTCAC(3)ATG | ||
| Concatenated domains | 13641 | 4547 | 97.4 | 99.6 | 91.8 | 84.4 | 87.2 | |||
| RaGD | 1ab | 296–20466 | 6723 | 83.7 | 84.4 | 84.4 | 80.0 | 76.4 | CTAAAC(215)ATG | |
| S | 20463–24542 | 1359 | 64.9 | 66.9 | 66.6 | 61.9 | 62.4 | CTAAAC(4)ATG | ||
| S1 | 20520–22652 | 711 | 56.7 | 58.7 | 57.9 | 53.6 | 54.5 | |||
| S2 | 22674–24535 | 621 | 74.0 | 76.3 | 76.4 | 71.4 | 72.2 | |||
| NS3a | 24542–25198 | 218 | 54.3 | 55.9 | 55.9 | 48.0 | 52.0 | CGTTAC(26)ATG | ||
| NS4a | 25200–25663 | 157 | 18.6 | 17.5 | 17.6 | NA | NA | CTTTGC(34)ATG | ||
| NS4b | 25294–25611 | 105 | 11.4 | 12.5 | 12.5 | NA | NA | |||
| E | 25654–25878 | 74 | 77.1 | 81.9 | 81.9 | 63.9 | 81.9 | CCAAAC(66)ATG | ||
| M | 25884–26624 | 246 | 78.1 | 79.9 | 80.3 | 78.1 | 78.8 | CTAAAC(3)ATG | ||
| N | 26635–27810 | 391 | 68.1 | 68.3 | 68.3 | 67.3 | 60.8 | CTAAAC(3)ATG | ||
| NS8 | 27821–28099 | 92 | 28.6 | 64.8 | 63.8 | NA | NA | CTAAAC(0)ATG | ||
| NS9 | 28123–28485 | 120 | 55.6 | 58.1 | 50.6 | NA | NA | CTTTAC(3)ATG | ||
| Concatenated domains | 13,674 | 4558 | 91.9 | 91.8 | 91.8 | 83.0 | 87.4 | |||
| NSP | Putative Functional Domain(s) | RsYN1 | RsYN2 | RsYN3 | RaGD | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Amino Acides Position in ORF1ab | Predicted Size (aa of Protein) | C-end Predicted Cleavage Site | Amino Acides Position in ORF1ab | Predicted Size (aa of Protein) | C-End Predicted Cleavage Site | Amino Acides Position in ORF1ab | Predicted Size (aa of Protein) | C-end Predicted Cleavage Site | Amino Acides Position in ORF1ab | Predicted Size (aa of Protein) | C-end Predicted Cleavage Site | ||
| NSP1 | Unknown | M1–A195 | 195 | VA│AP | M1-A195 | 195 | VA│SP | M1-A195 | 195 | VA│SP | M1-A195 | 195 | TA│PP |
| NSP2 | Unknown | A196–C896 | 701 | RC│GG | S196-S889 | 694 | RC│GG | S196-S889 | 694 | RC│GG | P196-S889 | 694 | RS│GG |
| NSP3 | ADRP, PL2 pro | G897–G2463 | 1567 | QG│SG | G890-G2453 | 1564 | AG│SG | G890-G2453 | 1564 | AG│SG | G890-G2462 | 1573 | NG│SG |
| NSP4 | Hydrophobid domain | S2464–Q2941 | 478 | LQ│SG | S2454–Q2931 | 478 | LQ│SG | S2454-Q2931 | 478 | LQ│SG | S2463-Q2940 | 478 | LQ│SG |
| NSP5 | 3CL pro | S2942–Q3243 | 302 | LQ│ST | S2932–Q3233 | 302 | LQ│ST | S2932-Q3233 | 302 | LQ│ST | S2941-Q3242 | 302 | LQ│SN |
| NSP6 | Hydrophobid domain | S3244–Q3519 | 276 | VQ│SK | S3234–Q3509 | 276 | VQ│SK | S3234-Q3509 | 276 | VQ│SK | S3243-Q3518 | 276 | VQ│SK |
| NSP7 | Replicase | S3520–Q3602 | 83 | LQ│SV | S3510–Q3592 | 83 | LQ│SV | S3510-Q3592 | 83 | LQ│SV | S3519-Q3601 | 83 | LQ│SV |
| NSP8 | Replicase | S3603–Q3797 | 195 | LQ│NN | S3593–Q3787 | 195 | LQ│NN | S3593-Q3787 | 195 | LQ│NN | S3602-Q3796 | 195 | LQ│NN |
| NSP9 | Replicase | N3798–Q3905 | 108 | LQ│AG | N3788–Q3895 | 108 | LQ│AG | N3788-Q3895 | 108 | LQ│AG | N3797-Q3904 | 108 | LQ│AG |
| NSP10 | RNA synthesis protein | A3906–Q4041 | 136 | VQ│SL | A3896–Q4031 | 136 | VQ│AL | A3896-Q4031 | 136 | VQ│AL | A3905-Q4040 | 136 | VQ│SL |
| NSP11 | Unknown (short peptide at the end of ORF1a) | S–D | 17 | A-D | 17 | A-D | 17 | S-N | 17 | ||||
| NSP12 | RdRp | S4042–Q4968 | 927 | LQ│AA | A4032-Q4958 | 927 | LQ│AA | A4032-Q4958 | 927 | LQ│AA | S4041-Q4967 | 927 | LQ│AA |
| NSP13 | Hel, NTPase | A4969–Q5565 | 597 | LQ│AG | A4959-Q5555 | 597 | LQ│AG | A4959-Q5555 | 597 | LQ│AG | A4968-Q5564 | 597 | LQ│AG |
| NSP14 | ExoN, NMT | A5566–Q6083 | 518 | LQ│GL | A5556-Q6073 | 518 | LQ│GL | A5556-Q6073 | 518 | LQ│GL | A5565-Q6082 | 518 | LQ│GL |
| NSP15 | NeudoU | G6084–Q6422 | 339 | LQ│AG | G6074-Q6412 | 339 | LQ│SG | G6074-Q6412 | 339 | LQ│SG | G6083-Q6421 | 339 | LQ│SG |
| NSP16 | 2’-O-MT | A6423–K6722 | 300 | S6413-K6712 | 300 | S6413-K6712 | 300 | S6422-K6723 | 302 | ||||
| Gene | Ka/Ks Ratio |
|---|---|
| NSP1 | 0.256 |
| NSP2 | 0.390 |
| NSP3 | 0.315 |
| NSP4 | 0.276 |
| NSP5 | 0.128 |
| NSP6 | 0.323 |
| NSP7 | 0.067 |
| NSP8 | 0.125 |
| NSP9 | 0.230 |
| NSP10 | 0.098 |
| NSP11 | 0.236 |
| NSP12 | 0.073 |
| NSP13 | 0.045 |
| NSP14 | 0.121 |
| NSP15 | 0.124 |
| NSP16 | 0.083 |
| S | 0.258 |
| ORF3a | 0.357 |
| ORF4a | 0.727 |
| ORF4b | 0.623 |
| E | 0.062 |
| M | 0.0691 |
| N | 0.230 |
| ORF8 | 0.276 |
| ORF9 | 0.843 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, N.; Luo, C.; Liu, H.; Yang, X.; Hu, B.; Zhang, W.; Li, B.; Zhu, Y.; Zhu, G.; Shen, X.; et al. Characterization of a New Member of Alphacoronavirus with Unique Genomic Features in Rhinolophus Bats. Viruses 2019, 11, 379. https://doi.org/10.3390/v11040379
Wang N, Luo C, Liu H, Yang X, Hu B, Zhang W, Li B, Zhu Y, Zhu G, Shen X, et al. Characterization of a New Member of Alphacoronavirus with Unique Genomic Features in Rhinolophus Bats. Viruses. 2019; 11(4):379. https://doi.org/10.3390/v11040379
Chicago/Turabian StyleWang, Ning, Chuming Luo, Haizhou Liu, Xinglou Yang, Ben Hu, Wei Zhang, Bei Li, Yan Zhu, Guangjian Zhu, Xurui Shen, and et al. 2019. "Characterization of a New Member of Alphacoronavirus with Unique Genomic Features in Rhinolophus Bats" Viruses 11, no. 4: 379. https://doi.org/10.3390/v11040379